 © Гасанов Вагиф Али оглы, руководитель ЦКП «Группа геномных технологий», кандидат медицинских наук.
© Гасанов Вагиф Али оглы, руководитель ЦКП «Группа геномных технологий», кандидат медицинских наук.
Область научных интересов: генетическая инженерия, экспрессия и модификация рекомбинантных белков.
Наиболее удобной, перспективной, модной и технологичной системой редактирования генома является система CRISPR. Количество публикаций растет с геометрической прогрессией, появляются различные вариации CRISPR. Однако возникший ажиотаж постепенно стал сходить на нет, что связано, прежде всего, с неожиданными результатами, которые возникают у исследователей в процессе работ. Всю суть проблем можно охарактеризовать следующим образом: одно лечим, другое калечим. Включая или выключая какой-либо ген, либо заменяя его, точно рассчитанная последовательность направляющей РНК вдруг находит себе мишень в ином месте генома и проявляться совершенно неожиданные результаты.
Суть метода CRISPR заключается в связывании выбранной геномной последовательности (мишени) с эффекторными белками (как правило, нуклеазами), посредством взаимодействия комлементарных РНК (спейсер) и ДНК (протоспейсер). Взаимодействие спейсера и протоспейсера опосредованного одиночной направляющей РНК (sgRNA или gRNA), ограниченной мотивом прилегающим к протоспейсеру (protospacer adjacent motif (PAM)). PAM представляет собой последовательность ДНК из 2–6 пар оснований, непосредственно следующую за последовательностью ДНК, на которую нацелен эффекторный белок системы CRISPR.
Наиболее популярным является использование системы CRISPR/Cas9, т.е. в качестве эффектроного белка выступает нуклеаза Cas9. А наиболее часто используемый фермент Cas9 это фермент Cas9 из Streptococcus pyogenes (обозначенной как SpCas9). Для CRISPR/SpCas9 PAM представляет собой последовательность 5'-NGG-3', где «N» это любое азотистое основание, за которым следуют два азотистых основания гуанина («G»).
Итак, в теории всё замечательно, однако в реальных экспериментах возникает большое количество несоответствий реального результата предполагаемому результату. Связано это с тем, что gRNA, оказывается способна связываться не только с целевым участком ДНК, но и с иными участками, нецелевыми (off-target).
Off-target связывание нуклеазы происходит из-за частичного, но достаточного совпадения с последовательностью-мишенью. Механизмы off-target связывания можно разделить на две основные формы: базовое несоответствие (или просто несоответствие mismatches) и выпуклое несоответствие (bulges). Mismatches это связывание gRNA с ДНК, не смотря на то, что из 20 аминокислот gRNA 3-5 аминокислот не соответствуют последовательности ДНК. Bulges это изменение вторичной структуры gRNA или ДНК протоспейсера, приводящей к тому, что за счет конформационного изменения (изгиба) несколько нуклеотидов не принимают участие во взаимодействии gRNA с ДНК, что приводит к неспецифическому слиянию молекул gRNA и ДНК.
Как показала работа, опубликованная еще в 2013 году [1] 99,96% сайтов, которые ранее считались уникальными мишенями Cas9 в экзонах человека, могут иметь потенциальные off-target эффекты, содержащие хотя бы одно несовпадение оснований в исходной последовательности.
Таким образом, у исследователя, работающего с линией клеток или с линейным животным, могут возникнуть трудности, которые весьма трудно преодолеть. В связи с этим появились различные информационные сервисы, позволяющие производить расчет потенциальных off-target для эталонных геномов. Но и они не всегда дают точный ответ, что ожидать в результате очередной модификации у сложного организма, так как не всегда могут предположить bulges и mismatches. Возникает вопрос, что же делать, когда применение CRISPR планируется не на линейном животном, а на целой популяции нелинейных организмов. Потенциальное количество проблем связанных с off-target возрастает во много раз. Именно поэтому огромный потенциал модификации генома CRISPR в качестве терапевтических препаратов, в том числе и для человека, пока еще не раскрыт.
Авторы комментируемой статьи разработали эффективный инструмент под названием CRISPRme, интегрирующий наборы данных генетических вариантов человека с ортогональными геномными аннотациями для прогнозирования и определения приоритетов off-target в исследовании. CRISPRme анализирует как однонуклеотидные варианты, так и вставки, учитывает истинные гаплотипы, допускает mismatches и bulges, а также подходит для анализа личного генома. Разработанный CRISPRme был применен для анализа результатов клинических исследований препарата для терапии серпо-клеточной анемии и β-талассемии на основе метода CRISPR, направленный на +58-kb интронный энхансер BCL11A.
Веб-продукт CRISPRme может интегрировать популяционные генетические варианты из наборов поэтапных индивидуальных вариантов (например, из 1000 Genomes Project (1000G) [2]), нефазированных индивидуальных вариантов (например, из проекта «Human Genetic Variation and Population» (HGDP) [3]) и вариантов на уровне популяции (например, из базы данных агрегации генома, (gnomAD) [4]), с учетом данных Exome Aggregation Consortium [5]. CRISPRme может принимать личные геномы для выявления и определения приоритетов частных off-target из-за вариантов, специфичных для одного конкретного человека. Также CRISPRme позволяет использовать до семи вариантов mismatches и до двух вариантов bulges как для спейсера, так и для протоспейсера по всему геному, включая PAM 5'-NNN-3'. Данный ресурс доступен в режиме он-лайн (crisprme.di.unlvr.it). Интерфейс CRISPRme представлен на рисунке 1.
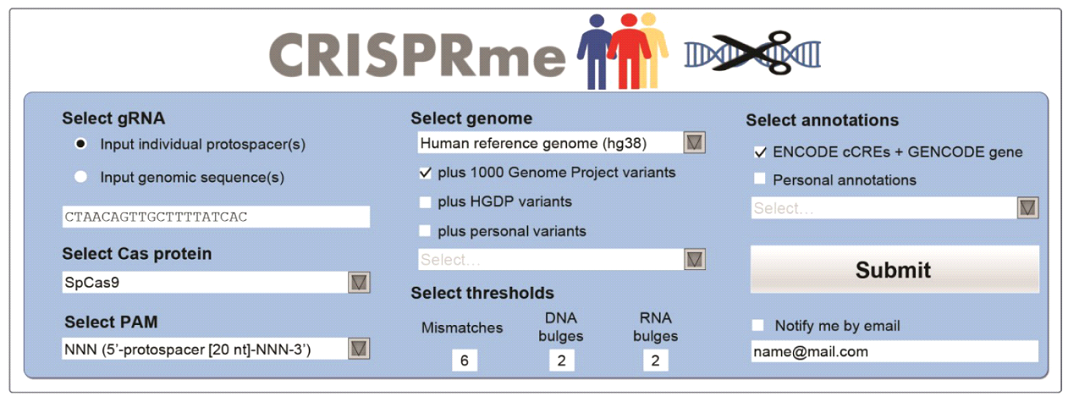
Используя CRISPRme авторы провели анализ результатов клинических исследований перспективного препарата для терапии серпо-клеточной анемии и β-талассемии и обнаружили, что верхний прогнозируемый off-target создается нереферентным аллелем, распространенным в популяциях африканского происхождения (rs114518452, частота минорного аллеля (MAF) = 4,5%), который вводит PAM для SpCas9. Авторы показали, что SpCas9 генерирует вставки с частотой более 9% в отредактированных клетках.
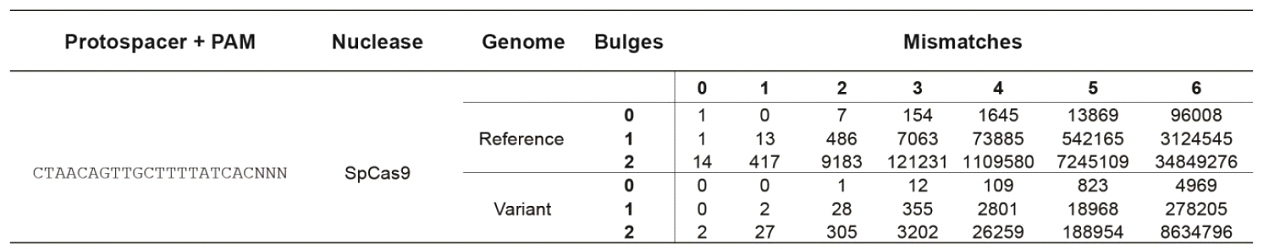
Были установлены все потенциальные off-target исследуемого спейсера, обнаруженные в эталонных или вариантных геномах, на основании их mismatches и bulges, что отражено на рисунке 2.
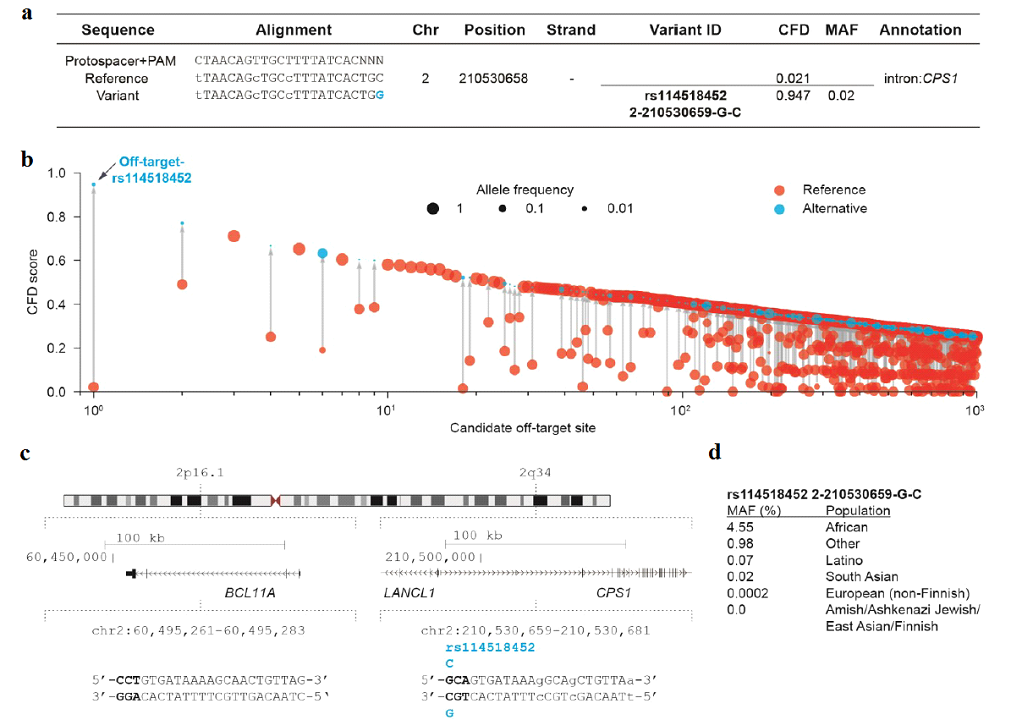
Авторами показана частота аллель-специфических off-target в суперпопуляциях и то, как генетическая изменчивость влияет на прогнозируемый потенциал расщепления с использованием показателей определения частоты разрезания (cutting frequency determination (CFD)). Были создан отчет по анализу связывания спейсера BCL11A-1617 нацеленного на мотив связывания GATA1 на +58 эритроидном энхансере BCL11A. В частности было показано связь с off-target повышение уровня фетального гемоглобина и отсутствие вазоокклюзионных эпизодов у некоторых пациентов на фоне терапии генно-модифицированными клетками, отредактированными с помощью SpCas9 и спейсера BCL11A-1617. Анализ CRISPRme показал, что предполагаемый off-target с наибольшим показателем CFD и наибольшим увеличением показателя CFD по сравнению с эталонным альтернативным аллелем находился в интронной последовательности CPS1 (см. рисунок 3), геномной мишени, подверженной общим генетическая изменчивость (модифицированная SNP с MAF ≥ 1%). Альтернативный аллель rs114518452-C генерирует последовательность 5’-TGG-3’ PAM (то есть оптимальную PAM для SpCas9) для потенциального off-target с 3 mismatches и оценка CFD (CFDalt 0,95), приближающаяся к оценке целевого сайта. Напротив, эталонный аллель rs114518452-G разрушает PAM до TGC, что заметно снижает прогнозируемый потенциал расщепления (CFDref 0,02). Альтернативный аллель rs114518452-C объединил MAF с общей частотой аллелей 1,33% в gnomAD v3.116, с MAF с частотой 4,55% у афро/афроамериканцев, 0,02% у европейцев и 0,00% у суперпопуляций Восточной Азии (см. рисунок 4).
Таким образом, авторы статьи не только разработали новый способ прогнозирования off-target в сложных суперпопуляционных исследованиях, но и доказали его эффективность, проведя анализ результатов, полученных при клинических исследованиях препарата на основе генно-модифицированных клеток.
В заключении хочется отметить не только объем и качество проведенных работ, а тот потенциал, который раскрывается перед разработчиками препаратов на основе редактирования генома, благодаря новой информационной платформе CRISPme.
Вне зависимости от того будет ли CRISPme развиваться в качестве самостоятельно сервиса, или сольется с каким-либо иным, но использование подобных сервисов должно стать рутинным для научных исследователей.
- Mali P. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology. (2013). doi:10.1038/nbt.2675. PMID 23907171
- Lowy-Gallego, E. et al. Variant calling on the GRCh38 assembly with the data from phase three of the 1000 Genomes Project. Wellcome Open Res 4, 50 (2019).
- Bergström, A. et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, (2020).
- Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
- Scott, D. A. & Zhang, F. Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med. (2017) doi:10.1038/nm.4377.
Новость подготовил
© Гасанов В.А.
27.02.2023