
 © Люпина Юлия Вячеславовна, ведущий научный сотрудник лаборатории биохимии процессов онтогенеза, доктор биологических наук. Область научных интересов: протеомика, структура и функции белков, контроль клеточного протеома, связывающие белки, макромолекулярные комплексы в развитии, эволюция белков.
© Люпина Юлия Вячеславовна, ведущий научный сотрудник лаборатории биохимии процессов онтогенеза, доктор биологических наук. Область научных интересов: протеомика, структура и функции белков, контроль клеточного протеома, связывающие белки, макромолекулярные комплексы в развитии, эволюция белков.
© Михайлов Виктор Сергеевич, главный научный сотрудник. Доктор биологических наук, профессор. Область научных интересов: репликация ДНК, репарация ДНК, ДНК-полимеразы, ДНК-связывающие белки, макромолекулярные синтезы в раннем развитии, репродукция вирусов.
Белки или протеины (от греческого «протос» – первый, важнейший) являются основным компонентом клеток и тканей и выполняют большинство функций в живых организмах. Однако технологии масштабных протеомных исследований отстали от методов изучения геномов. С момента получения первого полного эукариотического протеома (1), прогресс протеомики на основе метода масс-спектрометрии (2) позволил существенно продвинуться во всестороннем описании протеома человека (3-6). Однако до настоящего времени было мало сравнительных исследований протеомов у разных видов живых организмов (7, 8), что резко контрастирует с интенсивным изучением их геномов (9). В рецензируемой работе для изучения протеомов 100 организмов из различных таксонов авторы использовали усовершенствованный метод, в котором разделение пептидов выполняется чрезвычайно точной хроматографической системой. Всего авторы изучили два миллиона пептидов и идентифицировали более 340 000 белков в стандартизованных условиях. Эти результаты существенно расширяют представления научного сообщества о протеомике и позволяют по-новому взглянуть на функциональную организацию организмов, стоящих на разных ступенях эволюции.
Значительную часть общего протеома во всех царствах занимают белки, обеспечивающие гомеостаз и упаковку других белков, что подчеркивает важность поддержания правильной структуры белков для всех организмов. Медиаторы снабжения энергоресурсами также занимают заметное место в протеоме, хотя эти метаболические пути варьируют от фотосинтеза, метаболизма железа и серы до углеводного обмена. Как правило, белки и протеомы удивительно разнообразны у различных организмов, их можно анализировать и сравнивать на сайте, созданном авторами публикации - www.proteomesoflife.org.
Для выбора репрезентативных организмов по всему древу жизни, авторы учитывали наличие собранных геномов и доступность материала (например, тканей), а также взяли модельные организмы для сравнения. В итоге они выбрали 19 архей, 49 бактерий и 32 эукариотических организма — всего 100 различных видов. Авторы также дополнительно изучили 14 вирусов.
Авторы выяснили, что среди методов экстракции протокол in-StageTip (iST) (10) является наиболее универсальным и позволяет проводить автоматизированный анализ проб с высокой степенью воспроизводимости. Были использованы новейшие методы высокого разрешения, в том числе недавно разработанный метод чипирования (11, 12). Для всех прокариот авторы использовали однократную масс-спектрометрию (МС), а для образцов эукариот провели фракционирование (13). Хроматографический анализ на основе чипов в сочетании с большим массивом данных (более двух миллионов пептидов) идеально подходит для анализа нейросетью, который недавно стали использовать в протеомике на основе МС (14-16). Авторы разработали собственную рекуррентную нейросеть для анализа полученных данных и успешно протестировали ее на неизвестных ранее протеомах бактерий Bacteroides uniformis, Bacillus megaterium и Enterobacter aerogenes, используя результаты масс-спектрометрии. В итоге, свойства пептидов были успешно смоделированы in silico.
Для 100 организмов было идентифицировано 349 164 белка, которые четко разделялись по набору пептидов. Им соответствовали 1 136 558 учетных записей, 93% из которых были в TrEMBL-разделе базы данных UniProt (https://www.uniprot.org), содержащий белковые последовательности, предсказанные из геномов (17).
Данные авторов статьи значительно увеличивают количество экспериментально проверенных белков, особенно для бактерий и архей. Однако, даже у хорошо изученных модельных организмов удалось выявить много ранее неизвестных белков. База данных Swiss-Prot (версия 2019_03) включает 559 634 экспериментально проверенных белков всех изученных видов. После учета белков, которые были описаны ранее в базе PRIDE/ProteomeXchange (https://www.ebi.ac.uk/pride/archive/), авторы обнаружили дополнительно 803 686 белков, что более чем вдвое превышает общее количество белков, подтверждённых экспериментальными данными. Оценка глубины анализа протеомов была проведена авторами с использованием совокупности белков некоторых модельных организмов: дрожжей Saccharomyces cerevisiae, рыбы Danio rerio и хлопчатника Gossypium hirsutum, и оказалась выше, чем в проведенных ранее крупномасштабных исследованиях.
У прокариот авторы идентифицировали примерно половину всех белков, предсказанных на основе анализа геномов. Эукариоты обычно имеют более крупные геномы и, соответственно, авторами было выявлено большее количество белков. У некоторых видов показатели идентификации белков были низкими, что связано, по-видимому, с плохой аннотацией генома или протеома. В отличие от геномики и транскриптомики, данные протеомики позволяют провести прямой анализ конечного продукта экспрессии генов – белков (18, 19).
Определение гомологии белков представляет собой сложную биоинформатическую задачу, особенно трудно решаемую у плохо аннотированных организмов (20). Для сравнения всех видов, исследуемых авторами, использовали высококачественное предсказание гомологии из базы EggNOG 5.0 – базы ортологов. Авторы соединили данные по определенным белкам и пептидам с аннотациями и структурной информацией по ортологам из различных источников (17, 21-23) (рис. 1). Для нескольких видов авторы применили метод из пакета MaxQuant для количественной оценки интенсивности немеченных белковых фракций. Далее авторы выявили, как белки распределены по диапазону численности у различных организмов и рассчитали количество белков, которые вносят 90% вклада в общее количество белка по массе (рис. 2). При этом в графической базе данных возникает сетевая структура, с более чем 8 миллионами узлов (белки, пептиды, онтология генов и т. д.) и более 53,8 миллионами связей между ними. График можно построить для любой взаимосвязи между всеми этими узлами. Аналогичные запросы могут быть выполнены для целых биохимических путей, органелл или клеточных структур. Также можно исследовать взаимно зависимые пути, онтологию генов и их связь с нехарактерными белками (см. www.proteomesoflife.org).
Интересно, что белки, связанные с циклом протеома (трансляция, элонгация, упаковка и протеолиз) составляют примерно 10% протеома в живых организмах. Как и ожидалось, связанные с фотосинтезом белки присутствовали только в фотоавтотрофных организмах, таких как растения, водоросли, простейшие или цианобактерии (13 из 100 организмов). Фосфорилирование белков преимущественно наблюдается у эукариот, хотя некоторые бактерии и археи тоже используют эту модификацию, но их доля в протеоме существенно ниже, чем у эукариот. Около 38,4% идентифицированных белков не имели каких-либо функциональных аннотаций участия в биологических процессах, что было также у 22,9% из 100 наиболее распространенных белков каждого организма, а также для 10% функциональных доменов белков. Таким образом, данные статьи указывают на очень большое количество экспрессируемых белков с неизвестными функциями. «Темный протеом» интригует: его белки могут указывать на уникальные особенности эволюционного развития разных видов, которые могут иметь биологическое значение или представлять интерес для биотехнологии.
Cеквенирование новых геномов увеличивает число организмов, вовлеченных в исследование. В рецензируемой работе авторами сделан первый шаг к параллельному изучению протеомов. Выбрав виды из всех царств живых организмов, авторы создали большой набор изученных протеомов с большой выборкой экспрессирующихся белков. Такой подход позволяет выявить закономерности и особенности протеомов у близкородственных видов и делать сравнения с далекими родственниками всех таксономических уровней. Всем заинтересовавшимся исследователям предлагается использовать ресурс www.proteomesoflife.org.
Недостатком этого исследования является тот факт, что авторы изучали только часть типов клеток, тканей и биологических состояний, а также то, что глубина охвата протеома пока не является исчерпывающей. В работе почти не изучена посттрансляционная модификация белков и их эволюционное многообразие. Авторы предлагают направить усилия мирового научного сообщества на изучение протеомов других организмов в различных функциональных состояниях. Интеграция с геномикой, метаболомикой и другими сведениями, а также включение методов нейросетей видоспецифических библиотек позволило бы расширить познания за пределы модельных организмов для всего древа жизни.
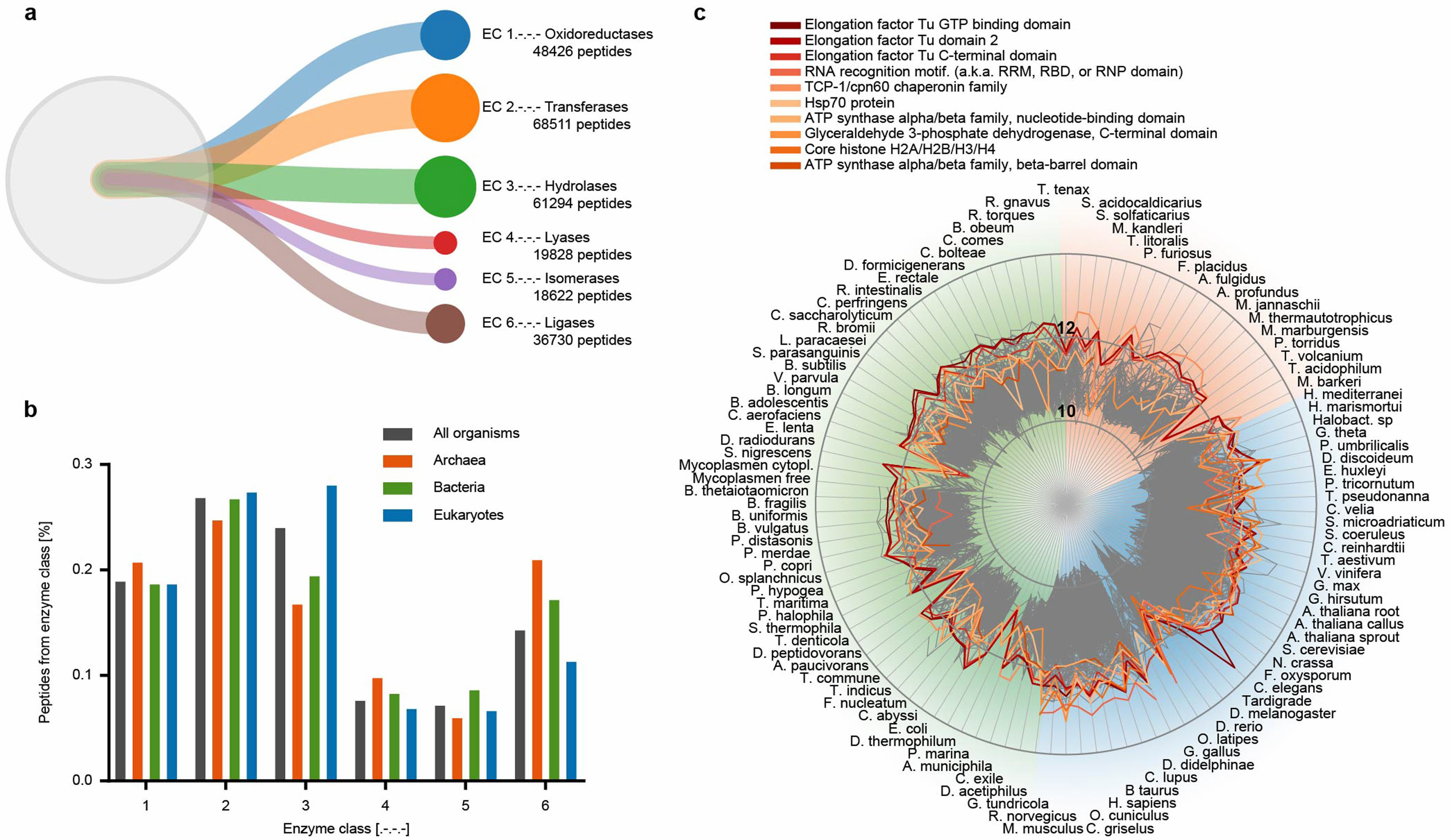
Рис. 1. Количественный анализ различных классов ферментов и их функциональных доменов по всему древу жизни.
а) вклад пептидов в 90% белковой массы у всех 100 изученных организмов с использованием Unipept (https://unipept.ugent.be/). Показаны пропорции каждого класса ферментов во всех исследованных организмах.
b) сравнение трех функциональных доменов в разных классах ферментов.
с) белки, составляющие 90% от суммарного белка во всех 100 исследованных организмах, аннотированные в соответствии с их известными функциональными доменами. Показаны наиболее распространенные функциональные домены белков у 100 организмов древа жизни.
Шкала представлена как log10.
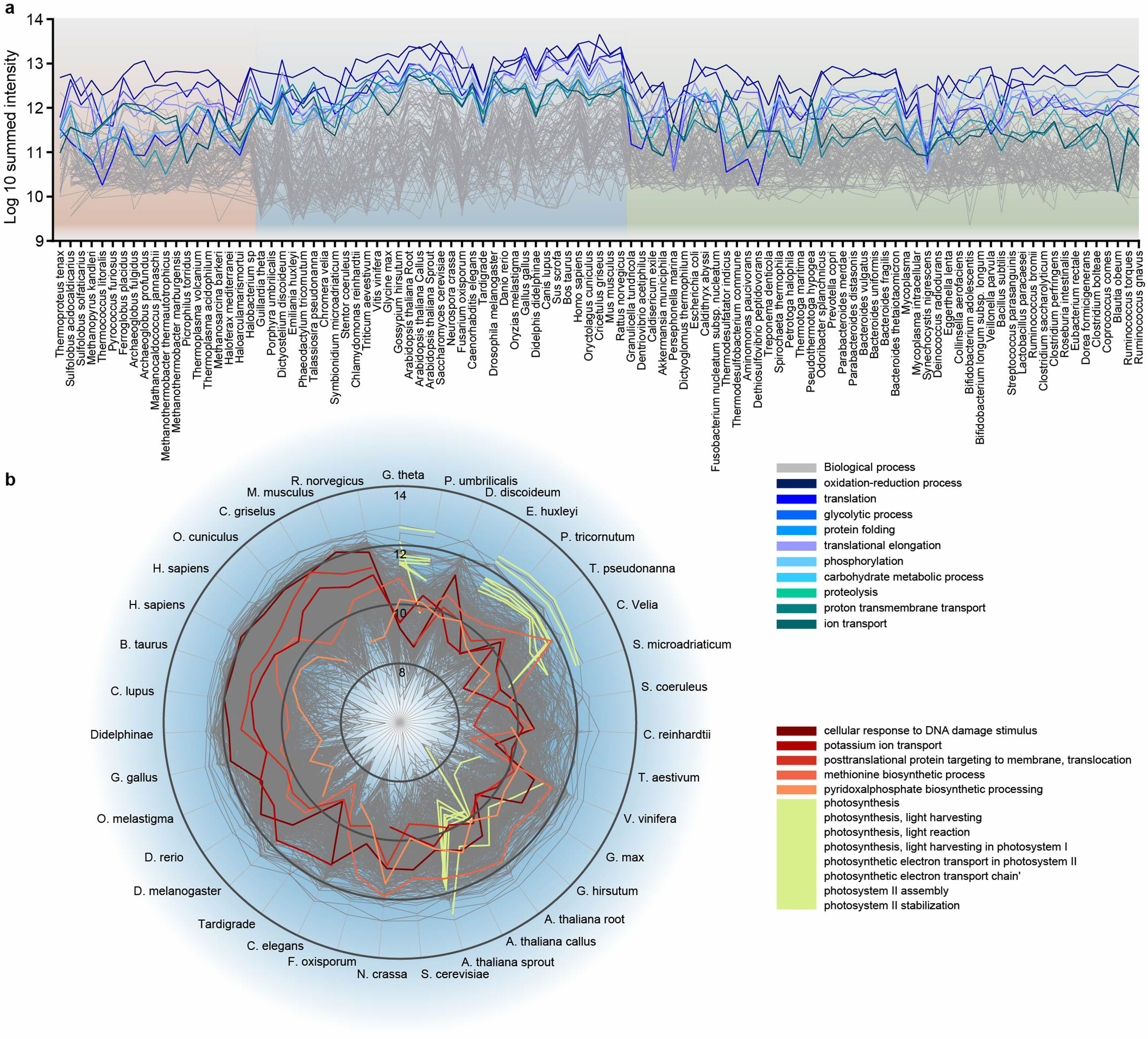
Рис. 2. Количественный анализ специфических биологических процессов у разных организмов древа жизни.
а) линейное изображение уровня экспрессии функциональных групп у 100 организмов;
b) количественный анализ участия белков в различных биологических процессах у эукариот. Белки аннотированы согласно их роли в биологических процессах с учетом уровня каждого белка в отдельных организмах.
REFERENCES
1. de Godoy, L. M. F. et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. 2008. Nature 455, 1251–1254.
2. Aebersold, R. & Mann, M. Mass-spectrometric exploration of proteome structure and function. 2016. Nature 537, 347–355.
3. Nagaraj, N. et al. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. 2012. Mol. Cell. Proteomics 11, M111.013722.
4. Kim, M.-S. et al. A draft map of the human proteome. 2014. Nature 509, 575–581.
5. Wilhelm, M. et al. Mass-spectrometry-based draft of the human proteome. 2014. Nature 509, 582–587.
6. Bekker-Jensen, D. B. et al. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. 2017. Cell Syst. 4, 587–599.
7. Weiss, M., Schrimpf, S., Hengartner, M. O., Lercher, M. J. & von Mering, C. Shotgun proteomics data from multiple organisms reveals remarkable quantitative conservation of the eukaryotic core proteome. 2010. Proteomics 10, 1297–1306.
8. Marx, H. et al. A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. 2016. Nat. Biotechnol. 34, 1198–1205.
9. Shendure, J. et al. DNA sequencing at 40: past, present and future. 2017. Nature 550, 345–353.
10. Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N. & Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. 2014. Nat. Methods 11, 319–324.
11. Geyer, P. E. et al. Plasma proteome profiling to assess human health and disease. 2016. Cell Syst. 2, 185–195.
12. De Beeck, J. O. et al. Digging deeper into the human proteome: a novel nanoflow LCMS setup using micro pillar array columns (μPACTM). 2018. Preprint at bioRxiv https://doi.org/10.1101/472134.
13. Kulak, N. A., Geyer, P. E. & Mann, M. Loss-less nano-fractionator for high sensitivity, high coverage proteomics. 2017. Mol. Cell. Proteomics 16, 694–705.
14. Zhou, X.-X. et al. pDeep: predicting MS/MS spectra of peptides with deep learning. 2017. Anal. Chem. 89, 12690–12697.
15. Tiwary, S. et al. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. 2019. Nat. Methods 16, 519–525.
16. Gessulat, S. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. 2019. Nat. Methods 16, 509–518.
17. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. 2019. Nucleic Acids Res. 47 (D1), D506–D515.
18. Munoz, J. & Heck, A. J. R. From the human genome to the human proteome. 2014. Angew. Chem. Int. Edn 53, 10864–10866.
19. Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. 2014. Mol. Cell. Proteomics 13, 2513–2526.
20. Altenhoff, A. M. et al. Standardized benchmarking in the quest for orthologs. 2016. Nat. Methods 13, 425–430.
21. The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. 2019. Nucleic Acids Res. 47 (D1), D330–D338.
22. Geer, L. Y. et al. The NCBI BioSystems database. 2010. Nucleic Acids Res. 38, D492–D496.
23. El-Gebali, S. et al. The Pfam protein families database in 2019. 2019. Nucleic Acids Res. 47 (D1), D427–D432.
Новость представлена © Ю. В.Люпиной и В. С. Михайловым
14.07.2020